
John Huddleston, Ph.D.
- Senior staff scientist in Bedford Lab and Bloom Lab
- Nextstrain developer
 @huddlej
@huddlej Google Scholar
Google Scholar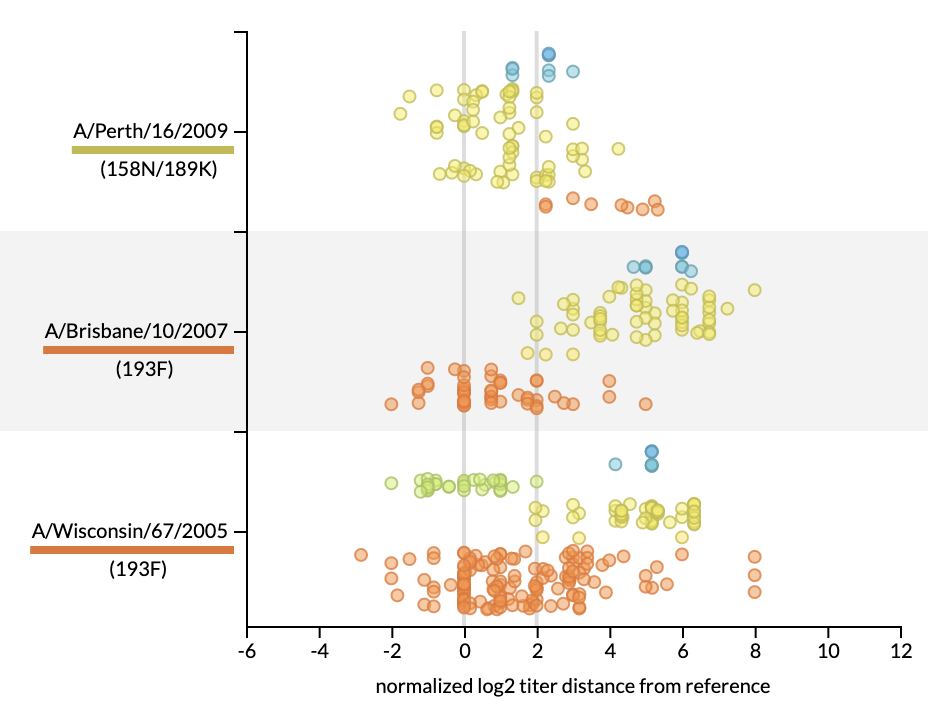
Joint visualization of seasonal influenza serology and phylogeny to inform vaccine composition
Lee and Hadfield, et al.
Seasonal influenza vaccines must be updated regularly to account for mutations that allow influenza viruses to escape our existing immunity. A successful vaccine should represent the genetic diversity of recently circulating viruses and induce antibodies that effectively prevent infection by those recent viruses. Thus, linking the genetic composition of circulating viruses and the serological experimental results measuring antibody efficacy is crucial to the vaccine design decision. Historically, genetic and serological data have been presented separately in the form of static visualizations of phylogenetic trees and tabular serological results to identify vaccine candidates. To simplify this decision-making process, we have created an interactive tool for visualizing serological data that has been integrated into Nextstrain’s real-time phylogenetic visualization framework, Auspice. We show how the combined interactive visualizations may be used by decision makers to explore the relationships between complex data sets for both prospective vaccine virus selection and retrospectively exploring the performance of vaccine viruses.
Published 22 Mar 2023 in Frontiers in Bioinformatics
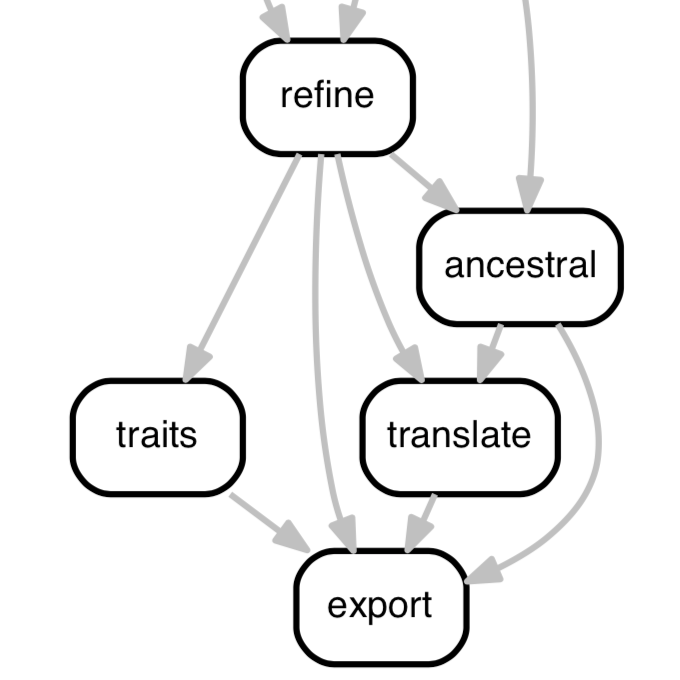
Augur: a bioinformatics toolkit for phylogenetic analyses of human pathogens
Huddleston, et al.
Real-time analysis of human pathogen evolution requires a diverse collection of bioinformatics tools to infer transmission dynamics and make recommendations to public health officials. These tools must scale with the number of samples and be flexible enough to adapt to a variety of questions and organisms. To meet these needs, we developed Augur, a bioinformatics toolkit designed for phylogenetic analyses of human pathogens.
Published 07 Jan 2021 in JOSS
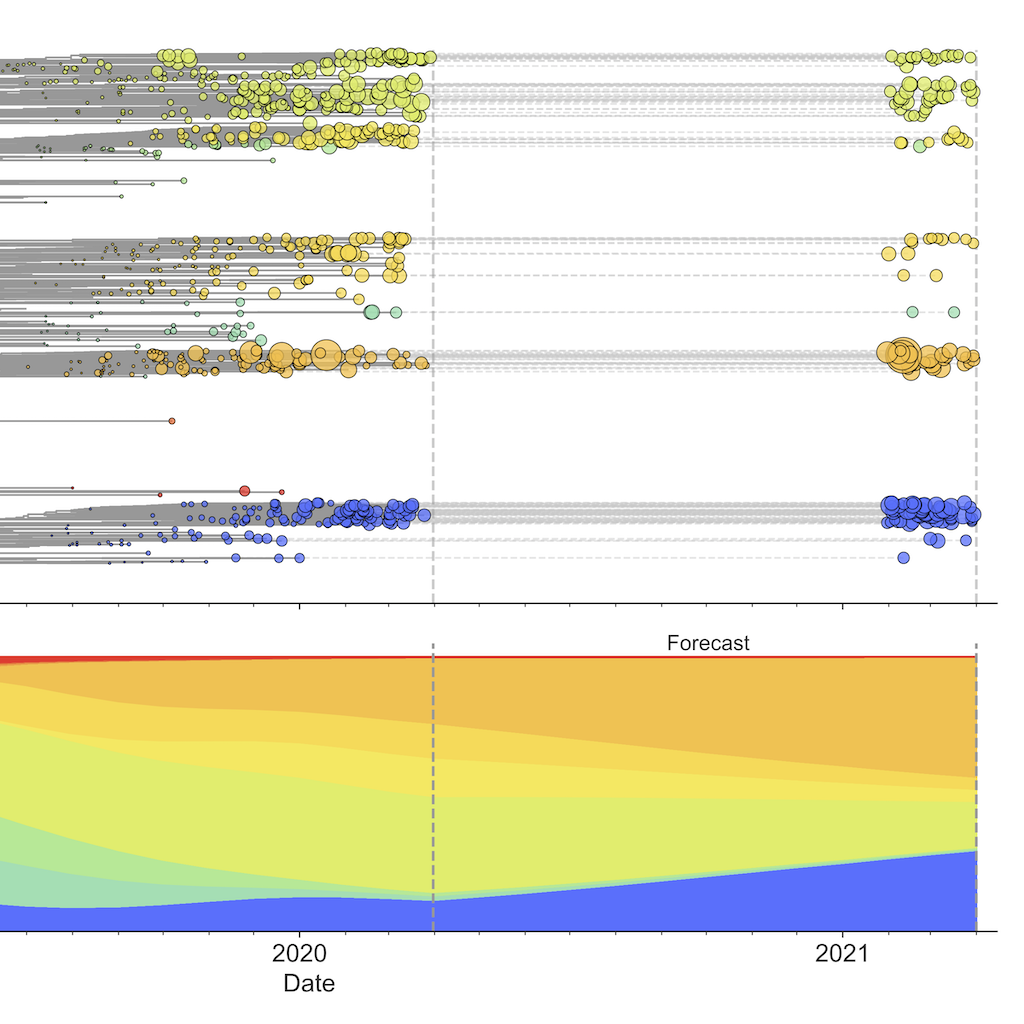
Integrating genotypes and phenotypes improves long-term forecasts of seasonal influenza A/H3N2 evolution
Huddleston, et al.
Rapid mutation of seasonal influenza’s hemagglutinin protein allows viruses to escape our adaptive immunity necessitating regular vaccine updates. We developed a novel framework that forecasts influenza evolution by combining fitness estimates from experimental assays and genomic data. We found that experimental measurements of antigenic drift provide more accurate forecasts than genomic data alone.
Read the full paper or the associated blog post.
Published 02 Sep 2020 in eLife
dms-view: Interactive visualization tool for deep mutational scanning data
Hilton*, Huddleston*, et al.
The high-throughput technique of deep mutational scanning (DMS) has recently made it possible to experimentally measure the effects of all amino-acid mutations to a protein. We developed an interactive visualization tool, dms-view, to enable dynamic exploration of DMS data in linked views with other site-level data and 3D protein structures.
Published 17 Aug 2020 in The Journal of Open Source Science
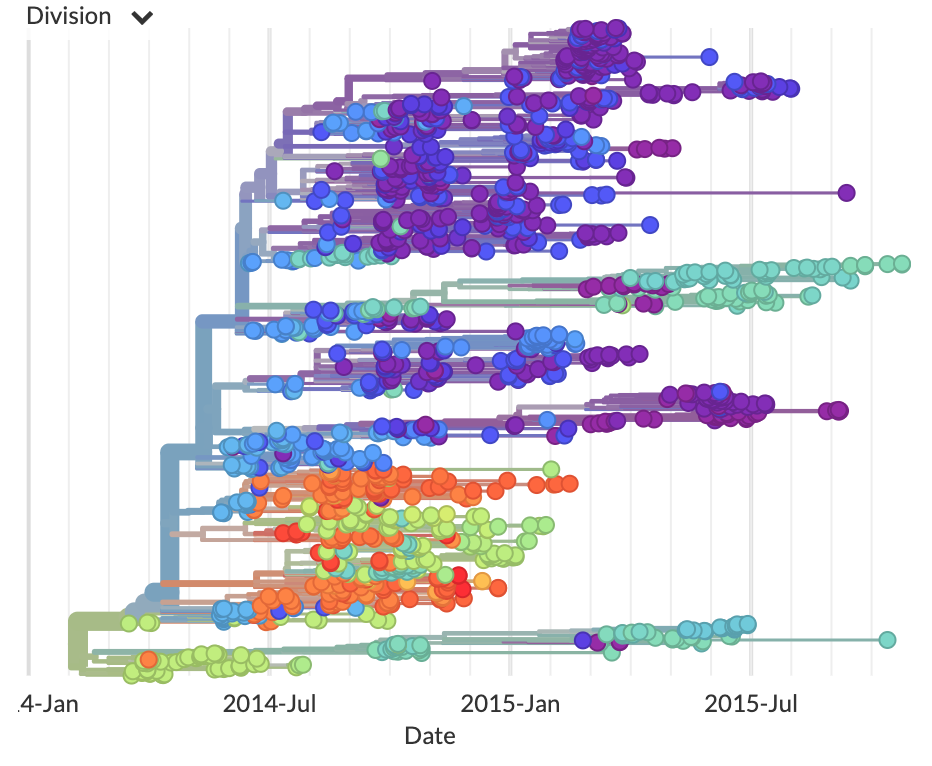
Nextstrain: real-time tracking of pathogen evolution
Hadfield, Megill, Bell, Huddleston, et al.
Understanding the spread and evolution of pathogens is important for effective public health measures and surveillance. To address this need, we developed Nextstrain, a database of viral genomes, a bioinformatics pipeline for phylodynamics analysis, and an interactive visualisation platform. Nextstrain compiles our current understanding into a single accessible location, publicly available for use by health professionals, epidemiologists, virologists and the public alike.
Published 01 Dec 2018 in Bioinformatics
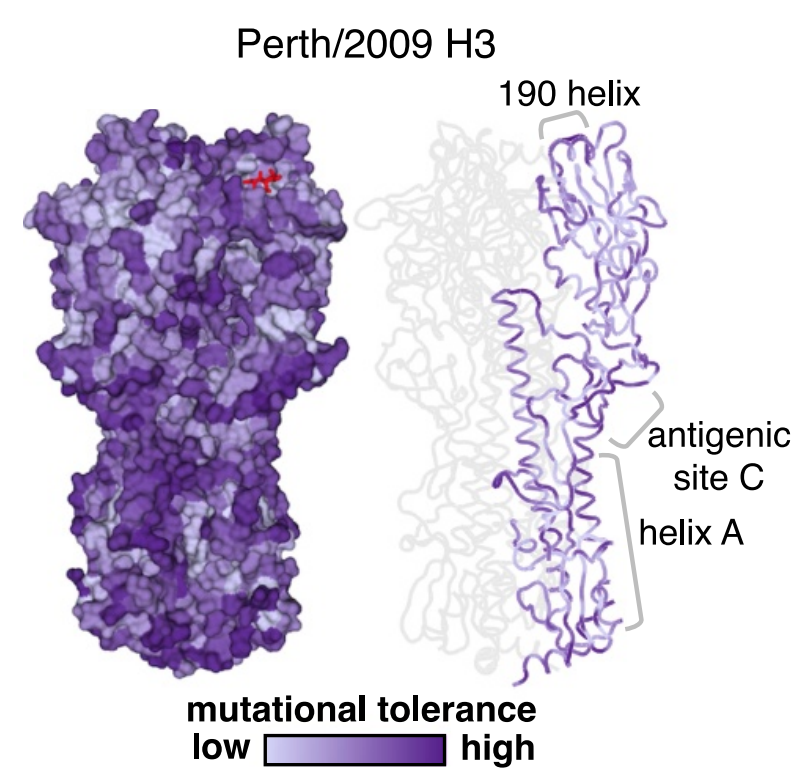
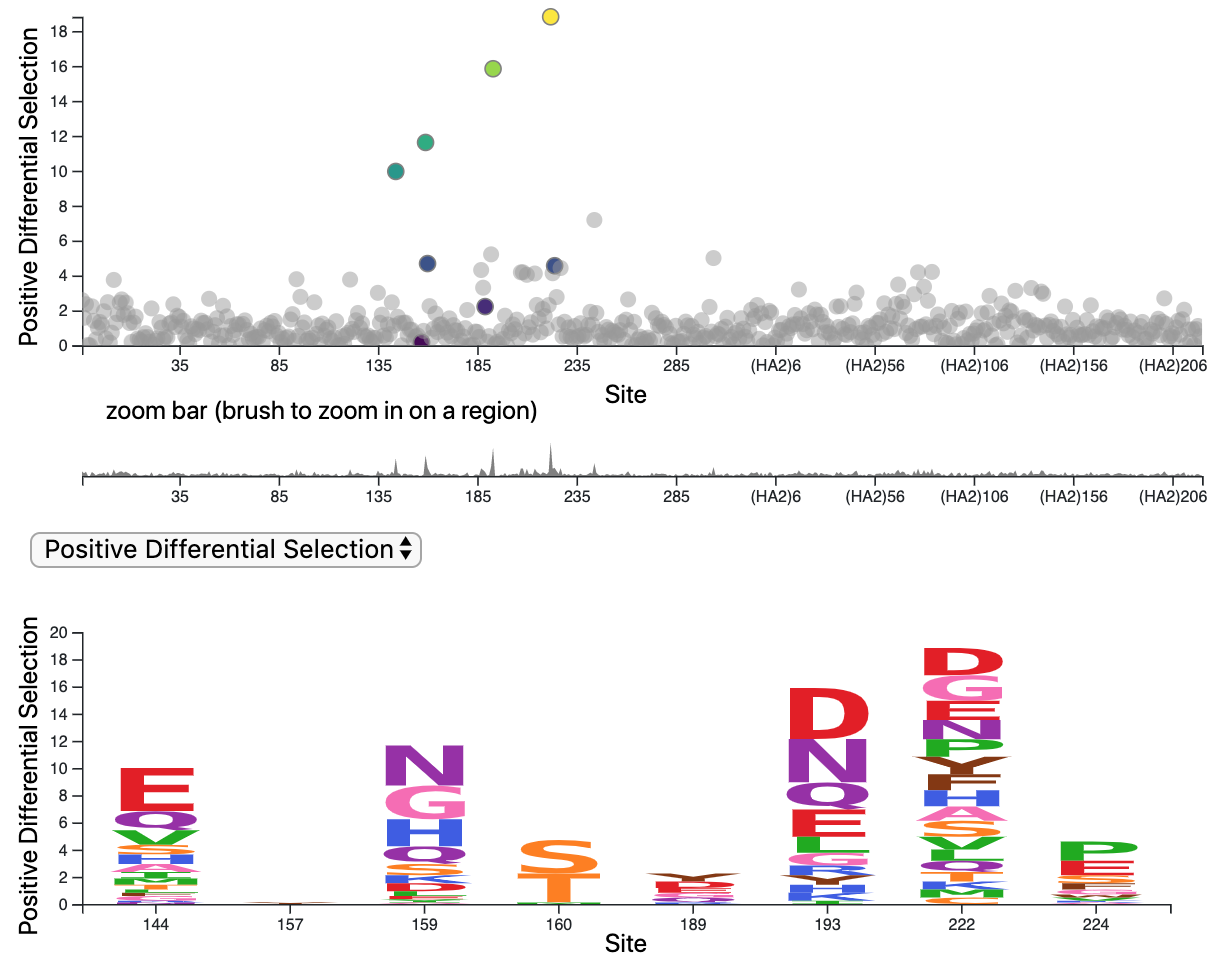
Deep mutational scanning of hemagglutinin helps predict evolutionary fates of human H3N2 influenza variants
Lee*, Huddleston*, et al.
Most efforts to understand the evolution of seasonal influenza A/H3N2’s surface protein, hemagglutinin (HA), focus on mutations that increase viral fitness by enabling escape from the immune system. We investigated the functional effects of all possible single amino-acid mutations to the HA of a single, recent H3N2 strain in laboratory conditions. We found that mutations with neutral or beneficial effects in the lab were often successful in nature, while mutations with deleterious effects were not.
Read the full paper or the associated blog post.
Published 13 Aug 2018 in PNAS